Make your Life Easier with the FREE Forms, ready for use in the Resource Center

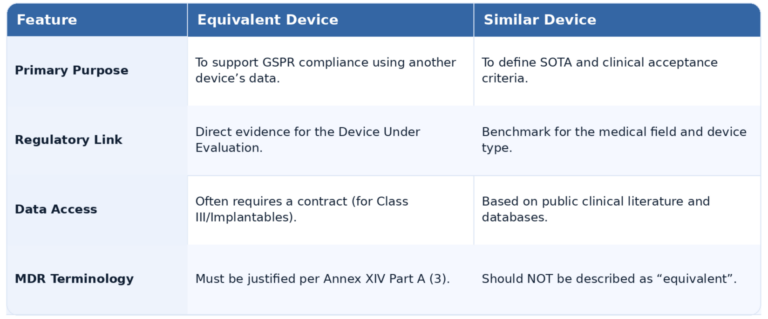
In the complex landscape of the European Union Medical Device Regulation (EU MDR 2017/745), the Clinical Evaluation Report (CER) is often described as the “beating heart of the Technical Documentation”. To ensure a successful submission, manufacturers must master two distinct but frequently confused concepts: Equivalent Devices and Similar Devices.
While they may sound alike, they serve different regulatory purposes and are subject to different evidentiary requirements.
Equivalent Devices: The Bridge to Clinical Evidence
The concept of “equivalence” is typically used when a manufacturer lacks sufficient clinical data for their own device and seeks to rely on data from another device to support compliance with General Safety and Performance Requirements (GSPR).
- When is it used? It is essential when one or more premarket clinical investigations have not been conducted for a new device.
- The “3-pillars” rule (must be equivalent together): Biological, technical, and clinical characteristics must be demonstrated as equivalent as a whole. Any differences must be justified as non-significant, typically through scientific rationale supported by testing, bench/biocompatibility evidence, and/or clinical literature.
- The “Access to Data” Hurdle: This is a critical determinant for acceptance by Notified Bodies. For Class III and implantable devices, the MDR generally requires a contractual agreement with the manufacturer of the equivalent device to ensure ongoing access to its technical documentation.
- Exemptions: For “Well-Established Technologies” (WET), a contract is not mandatory, provided equivalence is demonstrated and data access is justified.
- Future Outlook: Interestingly, a 2025 proposal to amend the MDR suggests removing the contract requirement to facilitate the use of clinical data from established devices and reduce barriers to claiming equivalence.
Similar Devices: Benchmarking the State of the Art (SOTA)
Unlike equivalent devices, similar devices are not used to “replace” data for your device. Instead, they are reviewed to understand the State of the Art (SOTA) and define the clinical context.
- Definition: Under the MDR, a similar device is generally understood as one falling under the same 4th level of the EMDN code.
- Why they matter: Reviewing similar device data allows manufacturers to:
- Identify clinical benefits and risks.
- Identify concerns or safety issues common to the device type.
- Define acceptance criteria: By looking at benchmark values (e.g., complication rates, adverse events, or efficacy measures) of comparable CE-marked devices, manufacturers can establish quantitative thresholds that their own device must meet
Search Strategies: Keeping them Separate
To satisfy Notified Body expectations, manufacturers should conduct at least two separate searches in their literature review:
- State of the Art Search: This focuses on similar devices, alternative treatments, and current clinical knowledge to establish a benchmark.
- Device Under Evaluation Search: This focuses specifically on your device and, if applicable, your equivalent devices to gather direct evidence of safety and performance.
Summary: The Key Differences
Conclusion
Manufacturers are highly recommended to include similar device data in their CER to provide a robust context for their device’s performance. However, you must refrain from using these terms interchangeably, as they have distinct meanings under the MDR. A clear, stratified analysis that respects these definitions will not only reduce follow-up questions from Notified Bodies but also build a more convincing rationale for your device’s safety and efficacy.