Medical Device Startup Support | LEXQARA:
Free ISO 13485 Templates, MDR Strategy & CE Marking

Medical Device Startup Support
Free ISO 13485 Templates + EU MDR Strategy + CE Marking Readiness
Build the right QA/RA & clinical foundations early — without wasting months guessing what “good” looks like.
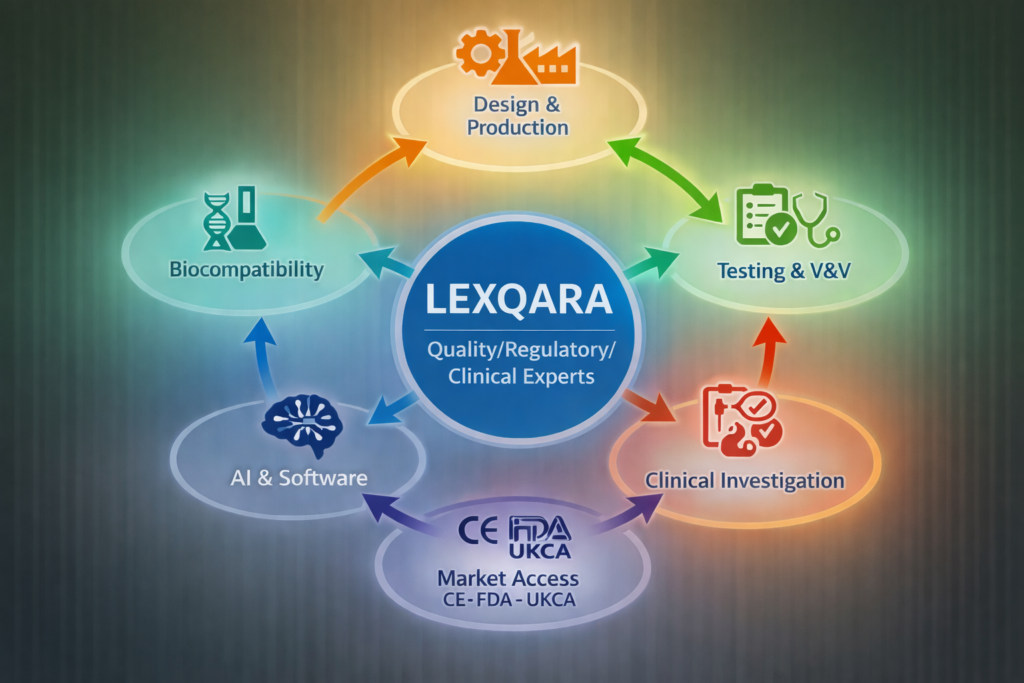
LEXQARA supports MedTech startups with a stage-based approach, backed by a qualified partner network for end-to-end execution:
design → production → testing → clinical investigation (if applicable) → regulatory submissions → market access (CE, FDA, UKCA).
Start today with:
- Free ISO 13485 forms & templates you can use immediately (not generic “dead” templates)
- A clear EU MDR 2017/745 roadmap aligned with your maturity and budget
- A hands-on partner + specialists when needed (biocompatibility, AI, clinical, labs, etc.)
The typical startup problem (and why it becomes expensive later)
Startups lose time and money when compliance starts too late or in the wrong order:
-
“We’ll do quality later”… until investors, hospital partners, or distributors request evidence
-
“We have templates”… but they don’t fit your product, so nobody uses them
-
“We need CE marking”… but traceability, records, and structure are missing
Our goal: help you build useful evidence now, so you don’t rebuild everything under pressure later.
One accountable partner — supported by specialists (A to Z)
LEXQARA combines Quality, Regulatory & Clinical expertise with a qualified partner network to deliver end-to-end support: design → production → testing → clinical investigation (if applicable) → CE marking, FDA 510(k)/PMA & UKCA.
LEXQARA coordinates your compliance journey and activates specialized partners when needed, so you keep one coherent plan and avoid fragmented vendors.
-
Product & industrialization support: design engineering, manufacturing setup, process validation
-
Testing: verification/validation, performance testing, electrical safety/EMC, software/cybersecurity (as applicable)
-
Biological evaluation: ISO 10993 strategy, toxicology, biocompatibility test coordination
-
Clinical: clinical strategy, clinical investigation setup (if required), study operations support
-
AI / software: AI lifecycle support, data/documentation readiness, alignment with applicable expectations
-
Market access: CE marking (EU MDR 2017/745), FDA 510(k) / PMA, UKCA
Result: a turnkey approach with clear accountability — LEXQARA coordinates, specialists execute where it matters.
One Partner, Three Stages: Templates → Strategy → Market Readiness
1) Start with foundations (free templates you can use immediately)
You don’t always need a full ISO 13485 QMS on Day 1.
But you do need the right records and habits from the start.
With LEXQARA, you receive free forms & templates, and we help you adapt them to your product and workflow so they become usable right away.
Typical outcomes:
-
A clean, startup-friendly documentation structure (simple, not bureaucratic)
-
Usable records for key topics (training, change control, CAPA, suppliers, document control…)
-
Clear guidance on what to document now vs. what can wait
Why this matters: these early records become future-proof evidence and will later integrate into a stronger, complete QMS.
2) Build your regulatory strategy (EU MDR 2017/745) — and prepare the future QMS
egulatory strategy is about making the right decisions early, so you build the right product and the right evidence (without overbuilding too soon).
LEXQARA helps you translate MDR expectations into a staged roadmap:
-
Product positioning & intended purpose/claims (what you build and how it is framed)
-
What to do now vs. later (risk management, clinical approach, PMS planning, technical documentation structure)
-
Prioritization aligned with your maturity level, budget, timeline, and target markets
Important: when the budget and timing are right, we can implement a real ISO 13485 QMS by building the procedures around the documents and records you already used and generated in Stage 1.
This avoids “starting from zero” and makes certification preparation faster and smoother.
3) Prepare for CE marking, ISO 13485 certification — and Notified Body readiness (end-to-end)
As you approach certification and submission, success depends on coherence: QMS + technical documentation + objective evidence must align and be audit-ready.
LEXQARA (and qualified partners when needed) can support the global pathway, including:
-
Design & development documentation (design control, risk management, V&V planning)
-
Production readiness and key quality elements (suppliers, controls, validations as applicable)
-
Testing strategy & coordination (performance/safety, software/cyber, biocompatibility…)
-
Clinical activities (if applicable): clinical strategy and clinical investigation setup/support
-
Regulatory documentation and technical file compilation for submission/audit readiness
Outcome: fewer last-minute surprises, stronger evidence, and higher confidence in front of a Notified Body.
Why startups choose LEXQARA
Free templates — but actually usable
Generic templates often remain unused. Ours are adapted to your workflow, so your team can apply them immediately and build robust evidence from day one.
Deep QA/RA/Clinical experience + pragmatic execution
Clear priorities, realistic deliverables, and alignment with real auditor expectations.
Reachable, hands-on support
Fast feedback loops and short cycles — built for startup pace.
Qualified partner network for end-to-end delivery
When your project needs specialized expertise (testing, biocompatibility, clinical, AI, industrialization…), you get access to trusted specialists, coordinated under a consistent compliance strategy.
Who this is for
A strong fit if you are:
-
A first-time MedTech founder needing clarity and structure
-
A startup building its first MDR roadmap and evidence plan
-
A team preparing ISO 13485 implementation over time
-
A company moving toward CE marking (and/or FDA/UKCA expansion)
FAQ — Medical Device Startup Support (EU MDR, ISO 13485, CE, FDA, UKCA)
Do I need ISO 13485 from the beginning?
Not always. But you should start early with the right records so you don’t rebuild everything later.
Can I use your templates even without a QMS?
Yes — they’re designed to be used immediately and to become the backbone of a future QMS.
What is the first step toward CE marking under EU MDR 2017/745?
Define intended purpose/claims, clarify positioning, then build a staged roadmap (risk, clinical, PMS, technical documentation structure).
When do we implement the full QMS with procedures?
When budget and timing align — and we build procedures around the documents/records you already generated in earlier stages.
Do you also support testing, biocompatibility, and clinical investigation?
Yes — via LEXQARA and qualified partners depending on what is applicable to your device.
Can you support FDA 510(k)/PMA and UKCA strategy?
Yes — we can align your roadmap for EU + US + UK as your market strategy expands.
What happens next?
LEXQARA works with international regulatory representatives (EU, Asia, Africa, Australia, the US, and South America) to facilitate your market access, and with qualified international consultants to support you throughout your regulatory process.