Quality Manual
Description:
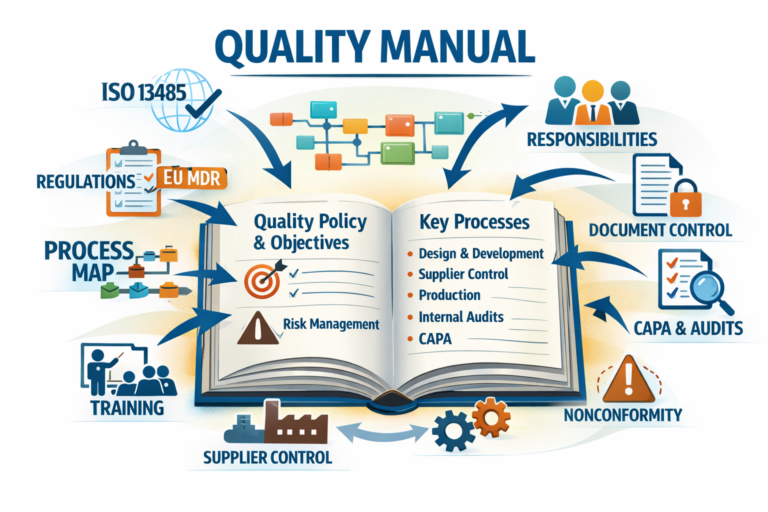
A Quality Manual is the top-level document of a Quality Management System (QMS). For medical device companies, it explains how your organization meets ISO 13485 requirements, defines the scope of your QMS, and shows how key processes work together to ensure product quality, patient safety, and regulatory compliance.
In practice, the Quality Manual is the starting point for employees, auditors, and notified bodies. It provides a clear overview of your QMS—without replacing your detailed procedures and work instructions.
What is a Quality Manual?
A Quality Manual is a controlled document that describes:
the scope of the QMS (sites, activities, product families, outsourced processes),
the standards and regulatory requirements applied (e.g., ISO 13485, applicable EU MDR obligations),
the structure of QMS documentation (how your procedures, SOPs, and records are organized),
the interaction of processes (often via a process map),
and the responsibilities and authorities for maintaining quality.
In other words, it’s the document that answers:
“How is our QMS built, what does it cover, and how do we control it?”
Why a Quality Manual matters for ISO 13485 and medical devices
For medical device manufacturers, the Quality Manual is essential because it supports a systematic, auditable approach to quality. A well-written manual helps you:
Demonstrate QMS scope and governance clearly during audits
Show the relationship between processes (design, purchasing, production, CAPA, etc.)
Improve internal alignment so teams understand how the system works
Reduce the risk of nonconformities caused by missing or inconsistent documentation
Support compliance expectations from customers, auditors, and notified bodies
A strong Quality Manual also simplifies onboarding and training, because it provides a single, structured reference to the QMS.
What a Quality Manual typically includes (medical device QMS)
A robust Quality Manual for ISO 13485 commonly includes:
1) QMS scope and applicability
Scope statement (what products/services/sites are covered)
Outsourced processes and how they are controlled
Any justified exclusions (where allowed)
2) Quality policy and objectives
Quality policy approved by top management
How quality objectives are established, monitored, and reviewed
3) QMS process architecture
Process map (core and support processes)
Process interactions (inputs/outputs and responsibilities)
4) Roles and responsibilities
Role of the organization (supplier, subcontractor, manufacturer, regulatory representative, importer, distributor, other)
Responsibilities and authorities for quality activities
Interfaces between functions (Quality, Regulatory, Operations, R&D, Supply Chain)
5) Overview of key QMS processes (with references)
High-level description of how your organization controls:
Document and record control
Risk management interface (how risk links to design, change, PMS, etc.)
Design and development (if applicable)
Supplier evaluation and purchasing controls
Production and service provision
Nonconformity control
CAPA (Corrective and Preventive Actions)
Internal audits
Management review
Training and competence management
…
The manual typically references the governing procedures rather than repeating them.
Quality Manual vs QSP vs SOP: what’s the difference?
Medical device QMS documentation is usually organized in levels:
Quality Manual (QM): the overview of your QMS (scope, structure, process interactions, responsibilities).
Quality System Procedures (QSPs): the system rules that define how major processes are managed (e.g., CAPA, audits, document control).
SOPs / Work Instructions: the step-by-step instructions to perform tasks consistently (e.g., incoming inspection, labeling checks).
Forms and Records: the evidence that the process was performed (e.g., training records, audit reports, DHR, CAPA forms).
Document Master List: the index showing current revision status, owners, and effective dates for all documents.
This structure ensures clarity, consistency, and audit readiness.
Needs:
Quality manual: Essential for compliance and operational efficiency.
- ISO 13485 outlines the framework that necessitates a comprehensive Quality Manual for medical device manufacturers.
- Resource Center offers various templates and guidelines to assist in the development of your Quality Manual.
- We offer support in drafting your Quality Manual, ensuring alignment with quality and regulatory requirements.
- Our expertise in quality management system implementation helps you establish effective procedures that are documented in your Quality Manual.
FAQ
Is a Quality Manual mandatory for ISO 13485?
A Quality Manual is mandatory under ISO 13485:2016. The standard requires a documented Quality Manual that describes the scope of your QMS and how your processes interact. Globally, many countries recognize ISO 13485 as the baseline for medical device QMS, while others apply their own QMS regulations that are closely aligned to it. In the EU, the MDR sets additional QMS obligations under Article 10(9), and Notified Bodies typically audit manufacturers against the current harmonized EN ISO 13485 standard and applicable MDR requirements, using ISO 13485 as the practical backbone of the QMS.
How long should a Quality Manual be?
Typically short: often 10–25 pages depending on complexity. Clarity is more important than length.
Should the Quality Manual include procedures?
No—usually it references procedures (QSPs/SOPs) rather than duplicating them.